- 스텔라라(우스테키누맙) 해외 바카라 사이트, 비교 임상 없이 FDA 첫 접수…승인과는 구분
- 비용·기간 절감→중견·중소 진입 확대
[더바이오 성재준 기자] 미국 식품의약국(FDA)이 최근 단일클론항체(mAb) 바이오시밀러(바이오의약품 복제약)에 대해 비교 임상시험 없이 처음으로 허가 심사 접수를 수용했다. 첫 적용 대상은 '스텔라라(Stelara, 성분 우스테키누맙)' 바이오시밀러다.
이달 확정된 최종 가이드라인에는 분석적 유사성과 면역원성 평가 등 비임상 자료의 비중을 확대하는 내용이 담겼다. 이 변화로 개발 비용·기간을 낮추고, 미국 내 바이오시밀러 도입 시점을 앞당길 수 있다는 평가다. 다만 이미 다양한 바이오시밀러를 신속하게 승인해온 유럽과 비교하면, 미국이 격차를 좁히기까지는 시간이 더 필요할 수도 있다.
30일 업계에 따르면, 해당 내용은 바이오시밀러 규제 전문가 사르파라즈 니아지(Sarfaraz K. Niazi) 미국 일리노이대학교 시카고 캠퍼스 교수가 공개한 발표를 통해 처음 밝혀졌다.
이번 사례는 아직 승인(approval)이 아닌 접수(acceptance) 단계다. 스폰서(개발사) 이름은 공개되지 않았다. 미국에서 이미 허가·출시된 우스테키누맙 바이오시밀러들과는 별개 사안이며, ‘분석 중심’ 심사가 실제 적용되기 시작했음을 시사한다.
FDA는 이달 ‘치료용 단백질 바이오시밀러 개발: 비교 분석평가 및 기타 품질 관련 고려사항(Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations)’ 최종본을 확정했다. 이 지침은 2019년 초안과 2015년 품질 가이던스를 대체하며, 구조·기능, 역가, 불순물, 제조공정 특성화 등 비교 분석평가와 품질(CMC) 자료를 중심으로 한 단계적 심사 원칙을 제시했다.
핵심은 고도의 분석적 유사성이 확보되면 비교 임상시험을 기본적으로 요구하지 않는다는 것이다. 면역원성과 약동학(PK)·약력학(PD) 등 임상 자료는 제품 특성에 따라 사례별로 판단하며, 불필요한 중복 임상은 최소화한다.
그간 FDA는 효과 동등성을 통계적으로 확인하기 위해 비교 임상시험을 요구하는 경우가 많았고, 수백~수천명 환자 모집이 필요해 시간·비용 부담이 컸다. 문턱이 낮아지면 중견·중소 기업의 진입이 늘고, 경쟁 확대를 통해 약가 인하 촉진과 환자 접근성 개선을 이끌 수 있다.
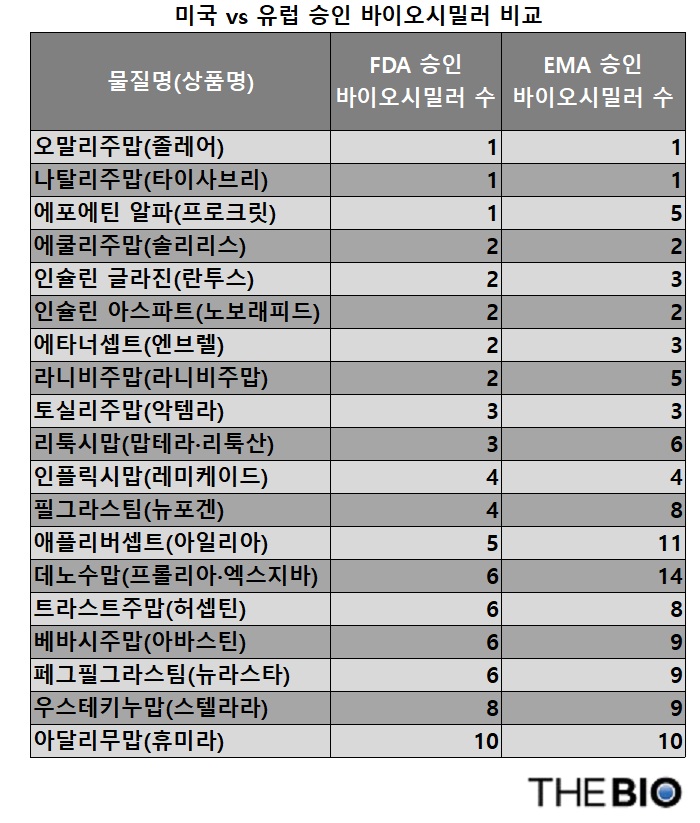
현 시점에서 바이오시밀러 승인 건수를 비교하면 유럽의약품청(EMA)쪽이 FDA보다 여전히 우위를 보인다. 데노수맙(denosumab, 프롤리아/엑스지바)에 대해 유럽과 미국이 14건 대 6건, 애플리버셉트(aflibercept, 아일리아)는 11 대 5, 리툭시맙(rituximab, 맙테라/리툭산)은 6 대 3, 라니비주맙(ranibizumab, 루센티스)는 5 대 2, 필그라스팀(filgrastim, 뉴포겐)은 8 대 4 등 주요 품목에서 격차가 난다.
반면 아달리무맙(adalimumab, 휴미라) 10 대 10, 인플릭시맙(infliximab, 레미케이드) 4 대 4처럼 균형을 이루는 경우도 있고, 우스테키누맙(ustekinumab, 스텔라라) 9 대 8처럼 비슷한 사례도 있다.
현재로선 미국이 유럽 수준에 도달하기까지는 시간이 더 필요할 것으로 보이지만 앞으로 가이드라인 변화가 상황을 바꿀 지 주목된다.